R-software multiMS-toolbox © 2012+
Introduction
multiMS-toolbox is a software toolbox to efficiently search for differences in mass-spectrometry samples from long-term experiments. It is supposed to have several runs for each sample. Then the software allows you to:
- match the appropriate peaks or peak clusters in the spectra of different runs and different samples and replace the peaks of the same isotope group by one group peak if required,
- select appropriate normalization method and run a principal component analysis (PCA, [Pearson 1901]) on processed data,
- group samples together and assign them the same shape or the same color in graphs, then draw the PCA scores plot (data samples plot) and loadings plot between each 2 of 3 dominant principal components, draw the 3D PCA scores plot,
- export and analyze each PCA component, draw the graphs of the most important PCA loadings and the most important peaks in the spectra,
- run analysis of variance (ANOVA) for each principal component and samples grouped according to same shape or color in graphs, draw the appropriate graphs,
- draw other graphs for later analysis, like the most important changes of absolute and relative intensities or areas of the peaks and peak clusters, output all the results to csv, txt, pdf (or tiff, png) files for later analysis.
Installation on MS Windows
The software is distributed in ZIP archive containing the „multiMS-toolbox" application folder. The toolbox main file is "multiMS-toolbox.R".
To use it you must first install the R-software system (https://www.r-project.org/) on your computer. To create 3D PCA plots and visualize match map of matched peaks, install also R-software packages rgl and ggplot2 (select Packages → Install package(s)… from the R gui menu). To check the characters in filenames, install also the R-software package stringi. To create average spectra plots, install also the R-software package reshape2. If you want to use normalization by best matching exponential line, install also the R-software package minpack.lm.
When you run the R-software, move to the directory, where you have installed the toolbox by setwd() command (to see the current directory, run the getwd() command from the R command line). For example, when multiMS-toolbox is installed in D:\multiMS-toolbox, run
> getwd()
[1] "D:/"
>setwd("D:/multiMS-toolbox")
And then load the toolbox by the source() command
> source("multiMS-toolbox.R")
You can assign the .RData extension to be automatically opened by the R for Windows GUI front-end and then run the R enviroment by double-click the 1blank.RData file (blank workspace) with the directory set to the current directory.
To run the demo examples for proteinaceous binders aging effect, move to the directory, where the example files are stored:
> setwd("examples")
> setwd("protbind")
And then run either of these commands:
> demoLowProteins1()
or according to selected normalization method (see Implemented functions for details)
> demoNormalizedLowProteins1()
> demoNormalizedLowProteins2()
> demoNormalizedLowProteins3()
For the full spectrum analysis available from version 2.0, you can also run
> demoFullSpectraNormalizedLowProteins1()
> demoFullSpectraNormalizedLowProteins2()
To run the demo examples for bacteria mass spectum, move to the directory, where the example files are stored:
>setwd("D:/multiMS-toolbox")
> setwd("examples")
> setwd("bacteria")
And then run the command
> demoHighProteins1()
or, when normalization is used, run
> demoNormalizedHighProteins1()
For the full spectrum analysis available from version 2.0, you can also run
> demoFullSpectraNormalizedHighProteins1()
> demoFullSpectraNormalizedHighProteins2()
All the outputs are printed and drawn to the R-GUI and stored to csv, pdf and txt files to the current directory.
Each of these demo functions only runs the core function runPCA having only two required parameters (lowMz, highMz) and several optional parameters. See the help for the given function in the Implemented functions section. The functions can be called from R command window, the passed parameters should include the name of the parameter and the value set. If the string value is set, then use quotes around the string, e.g.
> runPCA(csvfile="filesAll.csv", lowMz=900.0, highMz=2000.0)
Otherwise, you can specify all parameters in "config.R" file and specify only the path there:
> runPCA(paramsFile="configPeaksOnlyLowProteinsNormalize1.R")
If you find the next section too difficult, try our comon usecase examples for multiMS-toolbox.
Current version of multiMS-toolbox was successfully tested on Windows 7 64-bit with R 3.5.0 (64-bit environment).
Configuration file parameters
Parameters for file with ms peaks and spectrum information
csvfile – Excel’s csv file with the csvsep column separator. The default value is "filesAll.csv". The file should have column headers on the first line. The file should have at least columns:
- filesName – containing the names of data files to process.
- filesColorProperty – vector of string representations of graph colors for given files, the same string for two different files means, that its data points will be drawn with the same color.
- filesShapeProperty – vector of string representations of graph point shapes for given files, the same string for two different files means, that its data points will be drawn with the same shape.
- filesSpectrum – containing the names of spectrum files for given data files, this column is required either if findRealValuessForMissingPeaks is set to 1 or normalize is set to 1 or 3.
csvsep – the delimiter character between the columns in the read csvfile and in the written output files. The default value is ",".
csvdec – the decimal point character in read input files and written output files. For English, set it to ".". The default value is ".".
Crop spectrum parameters
lowMz – required parameter: the lowest used and displayed m/z value. No default value.
highMz – required parameter: the highest used and displayed m/z value.No default value.
Peak intensity and spectrum normalization parameters
normalize – input data normalization method:
- 0 – not normalized.
- 1 – peak intensities or areas are normalized by median of spectrum intensity ratios of each data spectrum to the template spectrum passed in the normalizedTemplateSpectrumFor1 parameter or to the first data sample spectrum.
- 2 – sum of all matched peak intensities or areas is normalized to the same value (sum of the first data sample). In case of full spectrum analysis, sum of the whole spectrum area is normalized to the same value.
- 3 – sum of the whole (cropped) spectrum area is normalized to the same value (sum of the first data sample) and the peak intensities are then appropriately rescaled. In case of full spectrum analysis, this option is equivalent to normalize=2.
- 4 – intensities are divided by best matching exponential line for given spectrum. This option is implemented only for full spectrum analysis in this version.
- 5 – each peak or spectrum intensity is scaled among the samples to have standard deviation equal to 1.
normalizedTemplateSpectrumFor1 – filename of the spectrum, which will be used as the template spectrum. If set to NULL then the first data sample spectrum will be used instead. The parameter is used only if normalize is set to 1 (normalization by median of spectrum intensity ratios of each data spectrum to template spectrum). The default value is NULL.
normalizeLowMz – the m/z start value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value. The default value is NULL, i.e. to be the same as lowMz.
normalizeHighMz – the m/z end value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value. The default value is NULL, i.e. to be the same as highMz.
useFullSpectra – 0 means run PCA on peaks, 1 means run PCA on full spectrum data and thus several other options like areaBased or deisotoped is then switched off. if this option is set, then the filesSpectrum column is required in the input csvfile. The default value is 0.
areaBased – which peak values use for the PCA:
- 0 – peak intensities.
- 1 – peak areas. Assuming Gaussian
distribution of peak intensities for each peak, areas are computed as
(full width at half maximum) . (peak intensity) .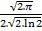
When used, fwhm and int columns are required in the data files.
- 2 – peak areas or partial peak areas. If partial areas are proportional to the whole area, PCA can be run only on partial peak areas (this holds for the Gaussian distribution for the intensities of each peak). When used, the area column is required in the data files.
deisotoping – 0 means all peaks are used, 1 means clusters are replaced by only one peak having the m/z value as first peak in the cluster. Peak intensity or area is then the sum of the processed intensities / areas of all the peaks within the cluster. When used, the deisotoping_grp column is required in the data files. The parameter is valid only if useFullSpectra is set to 0.
sn_cut – signal to noise ratio threshold. The default value is 0.0. When used with the value ≥ 0.0, the sn column is required in the data files. The parameter is valid only if useFullSpectra is set to 0.
maxDistance1 – the maximum m/z distance where peaks are treated as of the same m/z value. The default value is 0.3. The parameter is valid only if useFullSpectra is set to 0.
maxDistance2 – the maximum m/z distance where already matched groups of peaks (matched among several files by maxDistance1) will be treated as only one peak. The value is used only if it is higher than the maxDistance1 value. For more information about matching the peaks, see the Remarks section of the matchPeaks function. The default value is 0.51. The parameter is valid only if useFullSpectra is set to 0.
useRelativeMaxDistance – 0 means the maxDistance1 and maxDistance2
parameters are treated as absolute size of the interval to search, 1 means the maxDistance1 and maxDistance2
parameters are treated as multiplication coefficients. The absolute size of the interval to search is then
computed as maxDistance1 (or maxDistance2) multiplied by the m/z value of the peak.
For peaks with large m/z values, you can use for example:
useRelativeMaxDistance=1, maxDistance1=0.00015, maxDistance2=0.000255
The default value is 0. The parameter is valid only if useFullSpectra is set to 0.
findRealValuesForMissingPeaks – if set to 1 then for missing peaks (no match in given data file) their absolute intensity value is approximated from original spectrum file instead of setting them 0 intensity value (i.e. sn=0.0 intensity value for baseline subtracted intensity), or their area is approximated from intensity found in the original spectrum file and from minimum fwhm found between matched peaks of given m/z. if this option is set, then the filesSpectrum column is required in the input csvfile.The default value is 1. The parameter is valid only if useFullSpectra is set to 0.
fullSpectraDivide1MzBy - all the available spectrum data are interpolated from lowMz to highMz values and each 1 m/z is interpolated in fullSpectraDivide1MzBy intermediate values. The default value is 50. The parameter is valid only if useFullSpectra is set to 1.
fullSpectraMzTemplate:
- if set to any file name, than full spectra are interpolated at m/z points reads from the first column of given file restricted to the <lowMz, highMz> interval.
- if set to 1, than full spectra are interpolated at m/z points reads from the first sample spectrum file restricted to the <lowMz, highMz> interval.
- if set to -1, than full spectra are assumed to be already interpolated and only values inside the <lowMz, highMz> interval are used.
Experiment output parameters
label – character string to print in graphs and to use for file names (i.e. the name of the experiment). The default value is "".
numOfPCAComponents – number of principal components to show and to draw their graphs. The default value is 3.
itemsLabelAtMost – in the PCA scores plot this parameter specifies how large graphs will be plotted with labels assigned to each data point, in the PCA loadings plot the parameter specifies how many most extreme points will be plotted with their m/z values. If the PCA scores plot contains at most itemsLabelAtMost data points, then the data points will be plotted with their labels. Each label is a number representing the read order of given data point (data line in the original csvfile). In the PCA loadings plot, only to the first itemsLabelAtMost data points are plotted with their m/z values. The default value is 25.
legendColorPropertyLabel – the string showed in graph legends for grouping of samples based on colors - optional filesColorProperty column in the csvfile. The default value is "Colors".
legendShapePropertyLabel – the string showed in graph legends for grouping of samples based on shapes - optional filesShapeProperty column in the csvfile. The default value is "Shapes".
pdfFileWidthCm – the width of produced file outputs (in cm). The default value is 20.
pdfFileHeightCm – the height of produced file outputs (in cm). The default value is 20.
outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
dpi – dpi value for rasterized file outputs (tiff or png). Default value is 300.
Speed processing parameters
fast - 0 means compute and show all outputs, 1 means some long and time consuming but not essential outputs are omitted. Default value is 1.
Required format of peak and spectrum data files
MS Data files:
Data files, whose names are listed in the csvfile, should have the structure:
First line should contain the names of the data columns and each other line should contain data for one peak. There should be at least the columns mz and int. Optionally, there should be included the columns sn (when sn_cut has value ≥ 0.0), fwhm (when areaBased=1), area (when areaBased=2), or deisotoping_grp (when deisotoping=1). The column delimiter should be tab character.
MS Data file columns:
mz – m/z of the peak.
int – peak intensity after preprocessing.
sn – signal to noise ratio of the peak.
fwhm – full width at half maximum of the peak.
deisotoping_grp – either "None", if not a part of any peak cluster, or the number of the peak cluster.
area – area or partial area of the peak.
MS Spectrum file columns:
The spectrum is read from first two data columns (assuming no header line). In the first column there are m/z values, in the second column there are spectrum values (processed peak intensities). The spectrum data files needn’t to be sampled in the exactly same m/z points. The column delimiter should be tab character.
Implemented functions
runPCA function
Reads the data from the csvfile and normalizes them, matches the appropriate peaks, runs the PCA using Singular Value Decomposition, draws the graphs for components, computes ANOVA for each PCA component and group of data samples, exports the results.
Allowed parameters:
paramsFile – configuration file to load parameters from. The default values are overriden by values read from configuration file and those can be overriden by runPCA() function called with additional parameters. Use forward slashes to delimit path in Windows, e.g. "C:/multiMS-toolbox/examples/bacteria/configNotNormalized.R". The default value is NULL, i.e. no configuration file.
csvfile – Excel’s csv file with the csvsep column separator. The default value is "filesAll.csv". The file should have column headers on the first line. The file should have at least columns:
- filesName – containing the names of data files to process.
- filesColorProperty – vector of string representations of graph colors for given files, the same string for two different files means, that its data points will be drawn with the same color.
- filesShapeProperty – vector of string representations of graph point shapes for given files, the same string for two different files means, that its data points will be drawn with the same shape.
- filesSpectrum – containing the names of spectrum files for given data files, this column is required either if findRealValuessForMissingPeaks is set to 1 or normalize is set to 1 or 3.
csvsep – the delimiter character between the columns in the read csvfile and in the written output files. The default value is ",".
csvdec – the decimal point character in read input files and written output files. For English, set it to ".". The default value is ".".
lowMz – required parameter: the lowest used and displayed m/z value. No default value.
highMz – required parameter: the highest used and displayed m/z value.No default value.
normalize – input data normalization method:
- 0 – not normalized.
- 1 – peak intensities or areas are normalized by median of spectrum intensity ratios of each data spectrum to the template spectrum passed in the normalizedTemplateSpectrumFor1 parameter or to the first data sample spectrum.
- 2 – sum of all matched peak intensities or areas is normalized to the same value (sum of the first data sample). In case of full spectrum analysis, sum of the whole spectrum area is normalized to the same value.
- 3 – sum of the whole (cropped) spectrum area is normalized to the same value (sum of the first data sample) and the peak intensities are then appropriately rescaled. In case of full spectrum analysis, this option is equivalent to normalize=2.
- 4 – intensities are divided by best matching exponential line for given spectrum. This option is implemented only for full spectrum analysis in this version.
- 5 – each peak or spectrum intensity is scaled among the samples to have standard deviation equal to 1.
normalizedTemplateSpectrumFor1 – filename of the spectrum, which will be used as the template spectrum. If set to NULL then the first data sample spectrum will be used instead. The parameter is used only if normalize is set to 1 (normalization by median of spectrum intensity ratios of each data spectrum to template spectrum). The default value is NULL.
normalizeLowMz – the m/z start value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value. The default value is NULL, i.e. to be the same as lowMz.
normalizeHighMz – the m/z end value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value. The default value is NULL, i.e. to be the same as highMz.
useFullSpectra – 0 means run PCA on peaks, 1 means run PCA on full spectrum data and thus several other options like areaBased or deisotoped is then switched off. if this option is set, then the filesSpectrum column is required in the input csvfile. The default value is 0.
areaBased – which peak values use for the PCA:
- 0 – peak intensities.
- 1 – peak areas. Assuming Gaussian
distribution of peak intensities for each peak, areas are computed as
(full width at half maximum) . (peak intensity) .
When used, fwhm and int columns are required in the data files.
- 2 – peak areas or partial peak areas. If partial areas are proportional to the whole area, PCA can be run only on partial peak areas (this holds for the Gaussian distribution for the intensities of each peak). When used, the area column is required in the data files.
deisotoping – 0 means all peaks are used, 1 means clusters are replaced by only one peak having the m/z value as first peak in the cluster. Peak intensity or area is then the sum of the processed intensities / areas of all the peaks within the cluster. When used, the deisotoping_grp column is required in the data files.
sn_cut – signal to noise ratio threshold. The default value is 0.0. When used with the value ≥ 0.0, the sn column is required in the data files. The parameter is valid only if useFullSpectra is set to 0.
maxDistance1 – the maximum m/z distance where peaks are treated as of the same m/z value. The default value is 0.3. The parameter is valid only if useFullSpectra is set to 0.
maxDistance2 – the maximum m/z distance where already matched groups of peaks (matched among several files by maxDistance1) will be treated as only one peak. The value is used only if it is higher than the maxDistance1 value. For more information about matching the peaks, see the Remarks section of the matchPeaks function. The default value is 0.51. The parameter is valid only if useFullSpectra is set to 0.
useRelativeMaxDistance – 0 means the maxDistance1 and maxDistance2
parameters are treated as absolute size of the interval to search, 1 means the maxDistance1 and maxDistance2
parameters are treated as multiplication coefficients. The absolute size of the interval to search is then
computed as maxDistance1 (or maxDistance2) multiplied by the m/z value of the peak.
For peaks with large m/z values, you can use for example:
useRelativeMaxDistance=1, maxDistance1=0.00015, maxDistance2=0.000255
The default value is 0. The parameter is valid only if useFullSpectra is set to 0.
findRealValuesForMissingPeaks – if set to 1 then for missing peaks (no match in given data file) their absolute intensity value is approximated from original spectrum file instead of setting them 0 intensity value (i.e. sn=0.0 intensity value for baseline subtracted intensity), or their area is approximated from intensity found in the original spectrum file and from minimum fwhm found between matched peaks of given m/z. if this option is set, then the filesSpectrum column is required in the input csvfile.The default value is 1. The parameter is valid only if useFullSpectra is set to 0.
fullSpectraDivide1MzBy - all the available spectrum data are interpolated from lowMz to highMz values and each 1 m/z is interpolated in fullSpectraDivide1MzBy intermediate values. The default value is 50. The parameter is valid only if useFullSpectra is set to 1.
fullSpectraMzTemplate:
- if set to any file name, than full spectra are interpolated at m/z points reads from the first column of given file restricted to the <lowMz, highMz> interval.
- if set to 1, than full spectra are interpolated at m/z points reads from the first sample spectrum file restricted to the <lowMz, highMz> interval.
- if set to -1, than full spectra are assumed to be already interpolated and only values inside the <lowMz, highMz> interval are used.
label – character string to print in graphs and to use for file names (i.e. the name of the experiment). The default value is "".
numOfPCAComponents – number of principal components to show and to draw their graphs. The default value is 3.
itemsLabelAtMost – in the PCA scores plot this parameter specifies how large graphs will be plotted with labels assigned to each data point, in the PCA loadings plot the parameter specifies how many most extreme points will be plotted with their m/z values. If the PCA scores plot contains at most itemsLabelAtMost data points, then the data points will be plotted with their labels. Each label is a number representing the read order of given data point (data line in the original csvfile). In the PCA loadings plot, only to the first itemsLabelAtMost data points are plotted with their m/z values. The default value is 25.
legendColorPropertyLabel – the string showed in graph legends for grouping of samples based on colors - optional filesColorProperty column in the csvfile. The default value is "Colors".
legendShapePropertyLabel – the string showed in graph legends for grouping of samples based on shapes - optional filesShapeProperty column in the csvfile. The default value is "Shapes".
pdfFileWidthCm – the width of produced file outputs (in cm). The default value is 20.
pdfFileHeightCm – the height of produced file outputs (in cm). The default value is 20.
outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
dpi – dpi value for rasterized file outputs (tiff or png). Default value is 300.
fast - 0 means compute and show all outputs, 1 means some long and time consuming but not essential outputs are omitted. Default value is 1.
backupMemoryToDisk:
- if set to "SaveToDisk", it saves several memory object to files in current directory named as memory.*.rds for any faster future replots of these data.
- if set to "LoadFromDisk", it reads previously saved memory objects from files in current directory and replots them (including the 3D plots). This could speed up any previous data analysis. However, remember that the saved memory data are not valid for any kind of renormalization using multiMS-toolbox. To use different normalization method (normalize parameter) on data, do not use backupMemoryToDisk parameter with "LoadFromDisk" value and call the runPCA() function instead with appropriate new value of the normalize parameter.
- if set to NULL, no reading or saving memory from or to disk is allowed for memory objects.
Remarks:
For more information about processed operations, see the appropriate help of the other functions – for more information about matching the peaks see the Remarks section of the matchPeaks function, for more information about the ANOVA and the exported results, see the Remarks section of the showComponentsAndANOVA function.
init function
Fulfills the initial param structure.
Returns:
Returns the initial parameters structure.
readFilesCore function
Reads the spectrum peaks from all the files listed in the csvfile, equal or higher than given signal to noise parameter and normalizes the data, when normalize is 1 or 3.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- csvfile – Excel’s csv file with csvsep separator for columns. For the other help see the parameters section of the runPCA() function.
- csvsep – the delimiter character between the columns in the csvfile. The column delimiter in the read peak data files and spectrum files should be tab character.
- csvdec – the decimal point character in the read input files. For English, set it to ".".
- normalize – input data normalization method:
- 0 – not normalized.
- 1 – peak intensities or areas are normalized by median of spectrum intensity ratios of each data spectrum to the template spectrum passed in the normalizedTemplateSpectrumFor1 parameter or to the first data sample spectrum.
- 2 – sum of all matched peak intensities or areas is normalized to the same value (sum of the first data sample).
- 3 – sum of the whole (cropped) spectrum area is normalized to the same value (sum of the first data sample).
- 4 – spectrum is divided by best matching exponential line, this option is implemented only for full spectrum analysis.
- 5 – each intensity is scaled among the samples to have standard deviation equal to 1, this option is implemented only for full spectrum analysis.
- normalizedTemplateSpectrumFor1 – filename of the spectrum, which will be used as the template spectrum. If set to NULL then the first data sample spectrum will be used instead. The parameter is used only if normalize is set to 1 (normalization by median of spectrum intensity ratios of each data spectrum to template spectrum).
- lowMz – the lowest used m/z value.
- highMz – the highest used m/z value.
- normalizeLowMz – the m/z start value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value.
- normalizeHighMz – the m/z end value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value.
- areaBased – which peak values use
for the PCA:
- 0 – peak intensities.
- 1 – peak areas. Assuming Gaussian
distribution of peak intensities for each peak, areas are computed as
(full width at half maximum) . (peak intensity) .
When used, fwhm and int columns are required in the data files.
- 2 – peak areas or partial peak areas. If partial areas are proportional to the whole area, PCA can be run only on partial peak areas (this holds for the Gaussian distribution for the intensities of each peak). When used, the area column is required in the data files.
- deisotoping – 0 means all peaks are used, 1 means clusters are replaced by only one peak having the m/z value as first peak in the cluster. Peak intensity or area is then the sum of the processed intensities / areas of all the peaks within the cluster. When used, the deisotoping_grp column is required in the data files.
- sn_cut – signal to noise ratio threshold. The default value is 0.0. When used with the value ≥ 0.0, the sn column is required in the data files.
- findRealValuesForMissingPeaks – if set to 1 then for missing peaks (no match in given data file) their absolute intensity value is approximated from original spectrum file instead of setting them the 0.0 intensity value. For the other help see the parameters section of the runPCA() function. Required here for testing the presence of filesSpectrum in the csvfile and for normalization of the read values (when normalize is set to 1 or 3).
Returns:
Returns the structure with read peaks ($data), filenames ($files) and properties about each file ($filesProperties). If normalization by medians of spectrum intensity ratios or normalization by the whole (cropped) spectrum area occurs, also returns normalization coefficients ($spectrumNormalizeRatio).
Remarks:
When deisotoping is set to 1, peaks are read and the peak cluster is constructed from subsequent peaks having the same deisotoping_grp number. If the peak with the deisotoping_grp=None is read, then it is assumed that this peak doesn’t belong to the constructed peak cluster and is read as the separate (simple) cluster. If the peak with the different deisotoping_grp number is read, then the previous constructed cluster is finished and the construction of the new peak cluster is started.
When normalize is set to 1, spectrum values are interpolated in approximately <normalizeHighMz, normalizeLowMz> points and then the median of ratios of this values divided by the template spectrum interpolated values is selected as the normalization coefficient.
When normalize is set to 3, spectrum areas between normalizeLowMz and normalizeHighMz is computed. The ratios to the spectrum area of the first spectrum are selected as the normalization coefficients.
matchPeaks function
Matches
the peaks in the peaks read from different files. If no peak is found for given
m/z value in selected file, then the appropriate sn=0.0 intensity level (i.e. 0
intensity) or (sn=0.0 intensity level x min fwhm x ![]() ) value is used.
) value is used.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- normalize – input data normalization method:
- 0 – not normalized.
- 1 – peak intensities or areas are normalized by median of spectrum intensity ratios of each data spectrum to the template spectrum passed in the normalizedTemplateSpectrumFor1 parameter or to the first data sample spectrum.
- 2 – sum of all matched peak intensities or areas is normalized to the same value (sum of the first data sample).
- 3 – sum of the whole (cropped) spectrum area is normalized to the same value (sum of the first data sample).
- areaBased – which peak values use
for the PCA:
- 0 – peak intensities.
- 1 – peak areas. Assuming Gaussian
distribution of peak intensities for each peak, areas are computed as
(full width at half maximum) . (peak intensity) .
When used, fwhm and int columns are required in the data files.
- 2 – peak areas or partial peak areas. If partial areas are proportional to the whole area, PCA can be run only on partial peak areas (this holds for the Gaussian distribution for the intensities of each peak). When used, the area column is required in the data files.
- normalizeLowMz – the m/z start value of the spectrum normalization interval, valid in this function only if normalize is set to 2.
- normalizeHighMz – the m/z end value of the spectrum normalization interval, valid in this function only if normalize is set to 2.
- maxDistance1 – the maximum m/z distance where peaks are treated as of the same m/z value.
- maxDistance2 – the maximum m/z distance where already matched groups of peaks (matched among several files by maxDistance1) will be treated as only one peak. The value is used only if it is higher than the maxDistance1 value.
- useRelativeMaxDistance – 0 means the maxDistance1 and maxDistance2
parameters are treated as absolute size of the interval to search, 1 means the maxDistance1 and maxDistance2
parameters are treated as multiplication coefficients. The absolute size of the interval to search is then
computed as maxDistance1 (or maxDistance2) multiplied by the m/z value of the peak.
For peaks with large m/z values, you can use for example:
useRelativeMaxDistance=1, maxDistance1=0.00015, maxDistance2=0.000255 - findRealValuesForMissingPeaks – if set to 1 then for missing peaks (no match in given data file) their absolute intensity value is approximated from original spectrum file instead of setting them 0 intensity value (i.e. sn=0.0 intensity value for baseline subtracted intensity), or their area is approximated from intensity found in the original spectrum file and from minimum fwhm found between matched peaks of given m/z. if this option is set, then the filesSpectrum column is required in the input csvfile.
- csvsep – the delimiter character between the columns in the written output files.
- csvdec – the decimal point character in the written output files. For English, set it to ".".
- fast - 0 means compute and show all outputs, 1 means some long and time consuming but not essential outputs are omitted.
- namef - filename base for making output filenames.
data – the data vector with columns in a given order:
- m/z value
- peak intensity or the peak area, based on the areaBased and deisotoping parameter from the initial parameters structure when read.
- filename, from which was the peak read.
- fwhm – if areaBased is set to 1 or 2, full width at half maximum of the peak.
files – the names of all the files read, from which are the peaks matched. The parameter is required for assigning the found peak values to the appropriate files and for the determination of correct dimension of the constructed matched data vector.
spectrumFiles – the names of spectrum files for given data files, this column is required only if findRealValuessForMissingPeaks is set to 1.
spectrumNormalizeRatio – the normalization coefficients for spectrum files, this column is required only if findRealValuessForMissingPeaks is set to 1.
Returns:
Returns the m/z values ($mzVector) of all matched peaks and their intensities or areas ($matchedVectors) in every sample file. The m/z value is the m/z value of the peak with the highest intensity or area found in the sample files.
Remarks:
The peak matching algorithm first sorts all the peaks read from the files according their values (intensities or areas) and then tries to match the peaks (in decreasing order of their value) to peaks read from other files. It stores the m/z value of the selected peak and goes through all the peaks in the neighborhood of this peak searching for peaks that were not matched to any peak up to now (!) and that are at most in the maxDistance1. If any peak is found, it is marked as matched to current peak (processed) and its value is written to the column belonging to a file from which it was read. If there is more than one peak from one file in given m/z distance interval, then the highest value is stored, however both peaks are marked as matched (processed). If no peak from a file is found in the given m/z distance, then the 0 value is used (i.e. sn=0.0 value for the baseline subtracted intensities or areas). If findRealValuesForMissingPeaks is set to 1, then for missing peaks their intensity is approximated from original spectrum file for given m/z. If areaBased>0, their area is computed using the intensity and the minimum fwhm found among the already matched peaks of given m/z.
After first matching step, another peak matching step is made. Two groups of matched peaks (and all their matching peaks from corresponding data files) are set as equal, if the m/z values of the groups (i.e. the m/z of the peak with the highest value) differ by no more than maxDistance2.
After the second step all the matched (and normalized) peak intensities or areas and approximated values from unmatched data files are exported to the matched values csv file and to the match map csv file. In the former there are matched peak values for all m/z values and for all data files, in the latter there are 0/1 values (not matched/matched) for all m/z values and for all data files.
Note: The new parameter useRelativeMaxDistance (from version 1.01) allows to compute max distances either as fixed (useRelativeMaxDistance = 0, the maxDistance1 and maxDistance2 parameters are treated as absolute size of the interval to search) or relative to the m/z value of the peak (useRelativeMaxDistance = 1, the maxDistance1 and maxDistance2 parameters are treated as multiplication coefficients to the m/z value of the peak to obtain the absolute size of the interval to search).
plotMatchedVectors function
For matched peaks the matched values from each file are plotted to the graph saved to the file.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
- dpi – dpi value for rasterized file outputs (tiff or png).
- fast - 0 means compute all outputs, 1 means some long and time consuming but not essential outputs are omitted.
- namef - filename base for making output filenames.
mzVector – the vector of m/z values of the matched peaks to show the labels in the PCA loadings plot.
matchedVectors – the data points with the intensities from each normalized spectrum.
files – the names of all the files read, assuming one data point for one file.
Returns:
Doesn’t return any value.
readFilesCoreAndMatchFullSpectra function
Reads the spectrum data from all the files listed in the csvfile for the full spectrum analysis, interpolates them in the <normalizeHighMz, normalizeLowMz> interval and normalizes them according to the selected normalization method.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- csvfile – Excel’s csv file with csvsep separator for columns. For the other help see the parameters section of the runPCA() function.
- csvsep - the delimiter character between the columns in the csvfile. The column delimiter in the read spectrum files should be tab character.
- csvdec – the decimal point character in read input files and written output files. For English, set it to ".".
- normalize – input data normalization method:
- 0 – not normalized.
- 1 – peak intensities or areas are normalized by median of spectrum intensity ratios of each data spectrum to the template spectrum passed in the normalizedTemplateSpectrumFor1 parameter or to the first data sample spectrum.
- 2 – sum of all matched peak intensities or areas is normalized to the same value (sum of the first data sample).
- 3 – sum of the whole (cropped) spectrum area is normalized to the same value (sum of the first data sample).
- 4 – spectrum is divided by best matching exponential line, this option is implemented only for full spectrum analysis.
- 5 – each intensity is scaled among the samples to have standard deviation equal to 1, this option is implemented only for full spectrum analysis.
- normalizedTemplateSpectrumFor1 – filename of the spectrum, which will be used as the template spectrum. If set to NULL then the first data sample spectrum will be used instead. The parameter is used only if normalize is set to 1 (normalization by median of spectrum intensity ratios of each data spectrum to template spectrum).
- lowMz – the lowest used and displayed m/z value.
- highMz – the highest used and displayed m/z value.
- normalizeLowMz – the m/z start value of the spectrum normalization interval, valid only if normalize is set to 1 or higher value.
- normalizeHighMz – the m/z end value of the spectrum normalization interval, valid only if normalizeis set to 1 or higher value.
- fullSpectraDivide1MzBy – all the available spectrum data are interpolated from lowMz to highMz values and each 1 m/z is interpolated in fullSpectraDivide1MzBy intermediate values.
- fullSpectraMzTemplate:
- if set to any file name, than full spectra are interpolated at m/z points reads from the first column of given file restricted to the <lowMz, highMz> interval.
- if set to 1, than full spectra are interpolated at m/z points reads from the first sample spectrum file restricted to the <lowMz, highMz> interval.
- if set to -1, than full spectra are assumed to be already interpolated and only values inside the <lowMz, highMz> interval are used.
- fast - 0 means compute and show all outputs, 1 means some long and time consuming but not essential outputs are omitted.
Returns:
Returns the structure with read and interpolated spectrum data ($spectrumData), filenames ($files, $spectrumFiles) and properties about each spectrum file ($filesProperties). If normalization by medians of spectrum intensity ratios or normalization by the whole (cropped) spectrum area occurs, also returns normalization coefficients ($normalizeRatio).
Remarks:
When normalize is set to 1, spectrum values are interpolated in approximately <normalizeHighMz, normalizeLowMz> points and then the median of ratios of this values divided by the template spectrum interpolated values is selected as the normalization coefficient.
When normalize is set to 2 or 3, spectrum area between normalizeLowMz and normalizeHighMz is computed. The ratios to the spectrum area of the first spectrum are selected as the normalization coefficients.
The normalized spectra are stored into the files with the tab delimiter.
plotSpectraAveragedByColor function
In the full spectrum analysis (useFullSpectra==1) plots averaged spectrum for each group of spectrum samples (grouping based on color only). These graphs are also stored to a file.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- normalize – input data normalization method:
- 0 – not normalized.
- 1 – peak intensities or areas are normalized by median of spectrum intensity ratios of each data spectrum to the template spectrum passed in the normalizedTemplateSpectrumFor1 parameter or to the first data sample spectrum.
- 2 – sum of all matched peak intensities or areas is normalized to the same value (sum of the first data sample).
- 3 – sum of the whole (cropped) spectrum area is normalized to the same value (sum of the first data sample).
- 4 – spectrum is divided by best matching exponential line, this option is implemented only for full spectrum analysis.
- 5 – each intensity is scaled among the samples to have standard deviation equal to 1, this option is implemented only for full spectrum analysis.
- useFullSpectra – 0 means run PCA on peaks, 1 means run PCA on full spectrum data and thus several other options like areaBased or deisotoped is then switched off. if this option is set, then the filesSpectrum column is required in the input csvfile.
- lowMz – the lowest displayed m/z value in graphs.
- highMz – the lowest displayed m/z value in graphs.
- csvsep - the delimiter character between the columns in the written csv outputs.
- csvdec – the decimal point character in written output files.
- legendColorPropertyLabel – the string showed in graph legends for grouping of samples based on colors.
- pdfFileWidthCm – the width of produced file outputs (in cm).
- pdfFileHeightCm – the height of produced file outputs (in cm).
- outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
- dpi – dpi value for rasterized file outputs (tiff or png).
- fast - 0 means compute and show all outputs, 1 means some long and time consuming but not essential outputs are omitted.
- name - label base for making plot titles.
- namef - filename base for making output filenames.
mzVector – the vector of m/z values of the matched peaks to show the labels in the PCA loadings plot.
matchedVectors – the data points with the intensities from each normalized spectrum.
files – the names of all the files read, assuming one data point for one file.
filesColorProperty – vector of string representations of graph colors for given files, the same string for two different files means, that its data points will be drawn with the same color.
Returns:
Doesn’t return any value.
Remarks:
The averaged spectra are stored into the files with the tab delimiter.
plotPCAGraphs function
When the PCA is finished, having the matched peak intensities or areas from data files as data points, the function plotPCAGraphs draws XY graphs of scores (data points) for each two of three dominant principal components, 3D graph of scores (data points) for three most dominant principal components, XY graphs of loadings (important m/z values) for each two of three dominant principal components and bar plots for variance and cumulative variance explained by each principal component. These graphs are also stored to a file.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- legendColorPropertyLabel – the string showed in graph legends for grouping of samples based on colors.
- legendShapePropertyLabel – the string showed in graph legends for grouping of samples based on shapes.
- pdfFileWidthCm – the width of produced file outputs (in cm).
- pdfFileHeightCm – the height of produced file outputs (in cm).
- outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
- dpi – dpi value for rasterized file outputs (tiff or png).
- itemsLabelAtMost – in the PCA scores plot this parameter specifies how large graphs will be plotted with labels assigned to each data point, in the PCA loadings plot the parameter specifies how many most extreme points will be plotted with their m/z values. If the PCA scores plot contains at most itemsLabelAtMost data points, then the data points will be plotted with their labels. Each label is a number representing the read order of given data point (data line in the original csvfile). In the PCA loadings plot, only to the first itemsLabelAtMost data points are plotted with their m/z values.
- useFullSpectra – 0 means run PCA on peaks, 1 means run PCA on full spectrum data and thus several other options like areaBased or deisotoped is then switched off. if this option is set, then the filesSpectrum column is required in the input csvfile.
- name - label base for making plot titles.
- namef - filename base for making output filenames.
mzVector – the vector of m/z values of the matched peaks to show the labels in the PCA loadings plot.
files – the names of all the files read, assuming one data point for one file.
pr – the PCA structure containing the $x array with the PCA coordinates of each data point.
filesColorProperty – vector of string representations of graph colors for given files, the same string for two different files means, that its data points will be drawn with the same color.
filesShapeProperty – vector of string representations of graph point shapes for given files, the same string for two different files means, that its data points will be drawn with the same shape.
Returns:
Doesn’t return any value.
computeLargestDifferenceToDim function
For each data point (i.e. sample file) it determines the largest difference from any other data point (in PCA component1 and PCA component2 coordinate projection) and computes the delta value attributable to average matched m/z value (its peak intensity or area). This could help to determine the importance of the correct baseline values subtracted of the input data before multiMS-toolbox is used (should be smaller in the order of magnitude).
Allowed parameters:
files – the names of all the files read, assuming one data point from one file.
numDim – the number of dimensions for each data point.
pr – the PCA structure containing the $x array with the PCA coordinates of each data point.
Returns:
Doesn’t return any value.
plotAbsoluteValueAndRelativeValueChangeGraph function
Plots the graph showing the peaks where the intensities or areas changed a lot in the absolute value. Then plots the graph showing the relative value change (in percent). Both graphs are also stored to a file.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- useFullSpectra – 0 means run PCA on peaks, 1 means run PCA on full spectrum data and thus several other options like areaBased or deisotoped is then switched off. if this option is set, then the filesSpectrum column is required in the input csvfile.
- areaBased – which peak values use
for the PCA:
- 0 – peak intensities.
- 1 – peak areas. Assuming Gaussian
distribution of peak intensities for each peak, areas are computed as
(full width at half maximum) . (peak intensity) .
When used, fwhm and int columns are required in the data files.
- 2 – peak areas or partial peak areas. If partial areas are proportional to the whole area, PCA can be run only on partial peak areas (this holds for the Gaussian distribution for the intensities of each peak). When used, the area column is required in the data files.
- pdfFileWidthCm – the width of produced file outputs (in cm).
- pdfFileHeightCm – the height of produced file outputs (in cm).
- outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
- dpi – dpi value for rasterized file outputs (tiff or png).
- name - label base for making plot titles.
- namef - filename base for making output filenames.
dataMatched – the data points with the matched peaks and their intensities or areas and a vector of m/z values of the matched peaks.
Returns:
Doesn’t return any value.
showComponentsAndANOVA function
Exports the results for each PCA component to a csv file. Draws several graphs for each PCA component. Computes one-way or two way analysis of variance (ANOVA) for each group of data points (by color x shape property, by color property, by shape property). Draws ANOVA box-plot graphs for each selected PCA component and stores the text output to a file.
Allowed parameters:
params – initial parameters structure, where at most these parameters are used:
- numOfPCAComponents – the number of PCA components to show.
- csvsep - the delimiter character between the columns in the written output files.
- csvdec - the decimal point character in the written output files. For English, set it to ".".
- useFullSpectra – 0 means run PCA on peaks, 1 means run PCA on full spectrum data and thus several other options like areaBased or deisotoped is then switched off. if this option is set, then the filesSpectrum column is required in the input csvfile.
- lowMz – the lowest used and displayed m/z value.
- highMz – the highest used and displayed m/z value.
- legendColorPropertyLabel – the string showed in graph legends for grouping of samples based on colors.
- legendShapePropertyLabel – the string showed in graph legends for grouping of samples based on shapes.
- pdfFileWidthCm – the width of produced file outputs (in cm).
- pdfFileHeightCm – the height of produced file outputs (in cm).
- outputdev – the extension (type) of produced file outputs. Default value is "pdf" for PDF files, other available formats are for example "tiff" for uncompressed TIFF or "png" for PNG files.
- dpi – dpi value for rasterized file outputs (tiff or png).
- fast - 0 means compute and show all outputs, 1 means some long and time consuming but not essential outputs are omitted.
- name - label base for making plot titles.
- namef - filename base for making output filenames.
files – the names of all the files read, assuming one data point for one file.
pr – the PCA structure with data points transformed to new coordinates.
mzVector – the vector of m/z values of the matched peaks.
matchedVectors – the data points with the intensities or areas of the matched peaks.
filesColorProperty – vector of string representations of graph colors for given files, the same string for two different files means, that its data points will be drawn with the same color.
filesShapeProperty – vector of string representations of graph point shapes for given files, the same string for two different files means, that its data points will be drawn with the same shape.
Returns:
Doesn’t return any value.
Remarks:
For each component this function:
- exports the appropriate data for each component to a csv file.
- draws graphs showing most important peaks and their values of PCA loadings, PCA (real max – min value) x loadings.
- computes one-way or two way analysis of variance for each group of data points (by color x shape property, by color property, by shape property).
- draws ANOVA box-plot graphs for each selected PCA component.
- stores the text output of ANOVA to a txt file.
- saves all the plotted graphs to a file.
To the csv file, for each PCA component there are exported the data for each matched peak, not only those listed in the graphs. ANOVA box plots show minimum, 1st quartile, median, 3rd quartile, maximum, mean (red cross) and sometimes the outliers (circles).
Data columns of each PCA component csv file:
m/z – the m/z value of the matched peak.
center of PCA origin in old cords – center of data points subtracted from the cluster in the original coordinates (i.e. m/z of each matched peaks).
minPCAComponentX projected sample – the intensities or areas of matched peaks in a pure projected data point having the same PCA component coordinate as the data point with the smallest score (coordinate).
maxPCAComponentX projected sample – the intensities or areas of matched peaks in a pure projected data point having the same PCA component coordinate as the data point with the largest score (coordinate).
PCAComponent X loadings (not normalized) – vector of loadings (component base vector, in old coordinates) of given PCA component, not normalized to 0..1 interval.
PCAComponentX (real max-min value)*loadings – the influence of the matched peak (i.e. the PCA loadings multiplied by the change in the intensity or area between the minPCAComponentX projected sample and the maxPCAComponentX projected sample)
Existing minPCAcomponentX observation – the intensities or areas of matched peaks in the data point having the smallest PCA component score (coordinate).
Existing maxPCAcomponentX observation – the intensities or areas of matched peaks in the data point having the largest PCA component score (coordinate).
Functions for faster data processing
extractFilesPropertiesToCSVfile function
Lists all the files of given pattern and if the files have the pattern
(color)(concentration)-(sampleAge)(sampleRun).extension
[A-Za-z]*[0-9]* –[0-9]*[a-f].extension
then it creates the semicolon separated csvfile with the columns filesName, filesColor, filesAge, filesRun, and filesConcentrations. You can rename or copy the data from any listed column to the columns filesColorProperty, filesShapeProperty and to use them for selecting the color and shape of the data points in the PCA graph.
Allowed parameters:
pattern – files to process.
outputCSVfile – Excel’s csv file with the semicolon (;) separator.
csvsep – the delimiter character between the columns in the csvfile. The column delimiter in the read peak data files and spectrum files should be tab character.
Returns:
Doesn’t return any value.
Troubleshooting
-
I do not see any graphs.
Try to run examples first. If you see graph outputs and pdf outputs, try to process your files. Read the output in the text window in R. Sometimes the output warns you to install other packages to your R installation. The toolbox is tested on R version 3.4. Sometimes the error or warning written there could point you to the source of the problem. If the error occurs during reading the files, look for the typos in file names written to csv file and check if all your data files really exist in source directory. Check whether path names and filenames do not contain some national characters.
-
I have a lot of files to process and the output to the R text window is stuck.
Reading all the data files could take a long time. If you do not have a fast hard drive installed in the computer, try to avoid any other work with files on disk to speed up the operations. The text written to the R text window sometimes announce you that you could suppress some outputs by setting the fast parameter to 1 and speed up the computation of results. The PCA on large matrices could also take a long time. If problems still occur, try to run the computation on smaller set of data first. If the problem arises only with big data sets, try to run the computations on a computer with more memory. For big data sets use 64bit edition of R software. Before computation, you can also set more virtual memory to R (at least to finish the computations even on computers where not enough memory is installed). To assign 10000 Mb virtual memory to R, run command:
>memory.limit(size=10000)
References
Pearson, K. (1901) On lines and planes of closest fit to systems of points in space. Philosophical Magazine, 2: 559-572. doi: 10.1080/14786440109462720
License
This program, along with all associated documentation, is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation. However, if you find the software useful, please cite the relevant papers:
Hrdlickova Kuckova, S., Rambouskova, G., Hynek, R., Cejnar, P., Oltrogge, D., and Fuchs, R. (2015) Evaluation of mass spectrometric data using principal component analysis for determination of the effects of organic lakes on protein binder identification. J. Mass Spectrom., 50: 1270–1278. doi: 10.1002/jms.3699
Cejnar, P., Kuckova, S., Prochazka, A., Karamonova, L., Svobodova, B. (2018) Principal component analysis of normalized full spectrum mass spectrometry data in multiMS-toolbox: An effective tool to identify important factors for classification of different metabolic patterns and bacterial strains. doi: 10.1002/rcm.8110
Common usecase examples
If you found the documentation too detailed or difficult, try our common usecase examples for multiMS-toolbox.
Last modified: 30.07.2025